创新 • 创优 • 创见
电化学析氢反应 (HER)是发展绿色能源技术的重要组成部分,为高效、可持续地制氢提供了一种经济有效的方法。一直以来,铂基贵金属催化剂是优异的析氢催化剂,但高昂的价格和极度匮乏的资源限制了其广泛的应用。因此,发展低成本、高效的非Pt电解水析氢催化剂成为研究的热点。
单原子催化剂 (SACs) 最大限度地提高了金属原子的利用效率,实现了原子经济性。但是,表面自由能的增加使得只有一种单一金属位点的SACs倾向于团聚,从而导致性能显著下降。与单原子体系相比,电子结构可调的多原子催化剂能进一步提高催化剂的本征活性和稳定性。在多原子催化体系中,相邻原子间的强化学相互作用可有效地稳定单个物种,防止团聚,从而形成高度稳定的活性中心。
基于此,将载体上的金属原子分散到最小的团簇或双核结构中可以通过调节配体原子、配位数和结构畸变来调节电子结构。特别是,异核金属原子催化可以通过调节金属活性中心来优化活性、稳定性和选择性,这在单金属体系中是看不到的。因此,利用双原子催化剂的双原子金属核相互作用可最大限度地提高原子利用率和催化活性。然而,由于在苛刻的合成条件下缺乏原子尺度控制技术,并且难以确定准确的非晶结构和活性位点,异核双金属团簇的合成和表征仍然是一个巨大的挑战。

针对单一金属位点的单原子催化剂易于团聚,金属原子利用率低、性能下降等问题,山西大学晶态材料研究所范修军教授和张献明教授提出了采用相邻原子间的强化学相互作用稳定双金属物种,从而形成高稳定活性中心的思路。以W和Mo的多金属氧酸盐(POMs)为前驱体,调节活性组分W和Mo的含量,采用可控的水热自组装和化学气相沉积(CVD)氮化工艺,制备了具有高活性中心的W-Mo双金属原子中心催化剂(W1Mo1-NG)。
W1Mo1-NG催化剂具有接近于零的起始电压和24 mV的过电位,是迄今为止报道的最有效的非贵金属催化剂之一。DFT计算表明W-O-Mo-O-C结构中电子是离域的,这使得W-Mo双原子催化剂具有良好的H吸附行为,在全pH范围具有良好的电催化析氢活性。
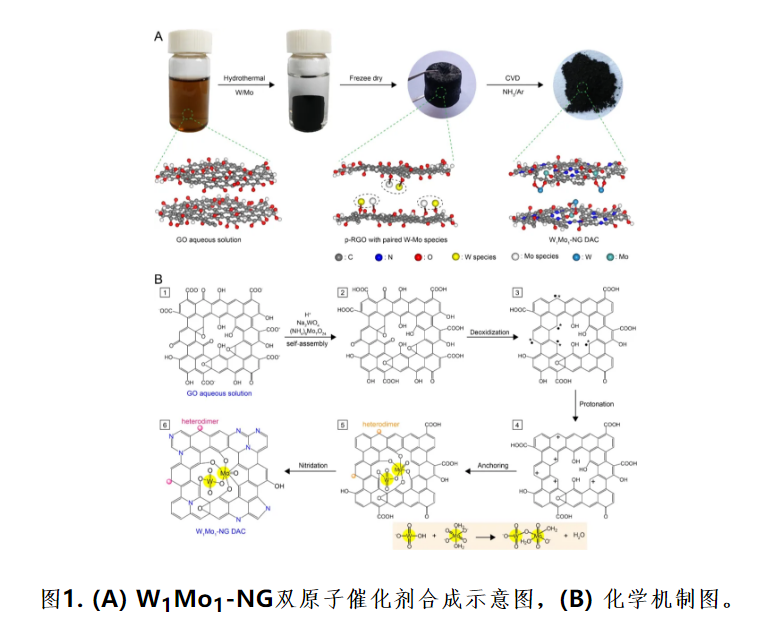
通过简单的水热和CVD的方法制备了W1Mo1-NG 双原子催化剂(图1A)。W1Mo1-NG形成机理是基于POMs自组装化学。具体地,将Na2WO4·2H2O和(NH4)6Mo7O24·4H2O添加到在pH 6.1 ~ 6.3的超声浴中的氧化石墨烯(GO) 悬浮液中图(1,图1B)。
随后,在水热过程中,发生了羧基阴离子的质子化(2,图1B),然后在GO上去除了质子化的羰基和环氧(3,图1B)。使离域的π电子系统和H+质子化以获得带正电的部分还原氧化石墨烯(p-RGO) (4,图1B)。在温和的酸性溶液中,质子化促进MoO42-片段中的Mo(VI)羰基转化为Mo羟基,最后转变为Mo水配体([MoO4(H2O2)]2-);而WO42-片段中的氢钨酸根阴离子([WO3(OH)]-) 保持四面体配位。
因此,可以通过脱水缩合反应将[WO3(OH)]-和[MoO4(H2O2)]2-自组装获得异核W-Mo物种。在聚集和重排之前,可以用质子化的p-RGO片作为反离子捕获并稳定异核W-Mo物种(5,图1B)。由于带负电的W-Mo二聚体阴离子和带正电的质子化p-RGO纳米片强烈偶联,O配位的W-Mo异二聚体可以在没有任何添加剂的情况下被静电吸引到p-RGO片上。
最后,通过氨退火制备均匀的W1Mo1-NG 双原子催化剂(6,图1B)。在CVD过程中,同时发生了p-RGO和N掺杂的进一步还原。W1Mo1-NG 双原子催化剂的成功合成归功于质子化p-RGO片与金属前驱体在弱酸性溶液中的结构不对称,其中质子化的p-RGO纳米片用作反离子可抑制WO42-和MoO42-碎片聚集成块状材料。
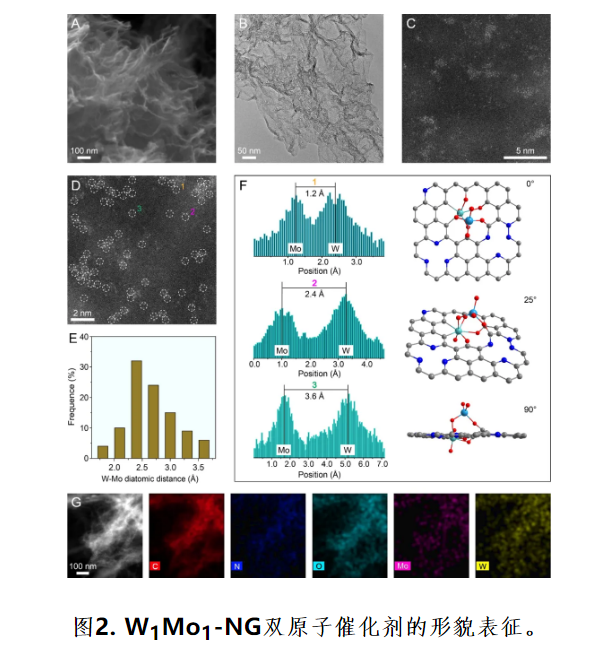
(A) 扫描电镜图,(B) 透射电镜图,(C-D) 高角环形暗场像-扫描透射电镜图,(E) 高角环形暗场像-扫描透射电镜图中的统计W-Mo双原子距离,(F) 在区域1、2和3中获得的强度分布图以及相应的示意图在视平面上显示了不同的W-Mo投影距离。(G) 元素映射图。
结合扫描电子显微镜(SEM),透射电子显微镜(TEM)和像差校正的高角度环形暗场扫描TEM (AC HAADF-STEM)图像给出了所制备催化剂的精细结构。图2A-B所示W1Mo1-NG有丰富的褶皱,可作为锚定位点来稳定金属物种。AC HAADF-STEM图像 (图2C) 呈现出高密度的小亮点,证实了W-Mo原子均与分布在氮掺杂石墨烯 (NG) 上。图2D-F所示成对的W-Mo原子分布在NG上。图2G所示C、N、O、Mo和W元素均匀的分布于NG上。
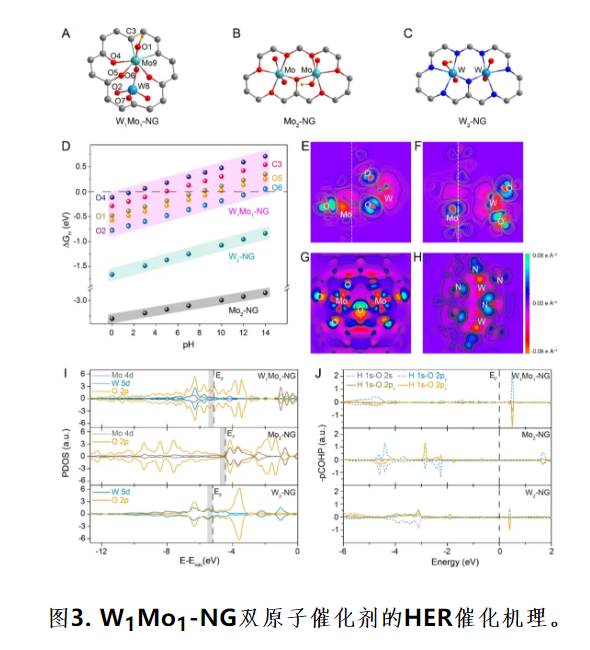
(A) W1Mo1-NG,(B) Mo2-NG和 (C) W2-NG优化的几何结构和可能的H吸附活性位点。(D) H吸附自由能图。(E-F) W1Mo1-NG,(G) Mo2-NG和 (H) W2-NG的差分电荷密度图。W1Mo1-NG,Mo2-NG和W2-NG模型的(I)投影态密度,(J)晶体轨道哈密顿布居数。
利用DFT计算对催化剂在全pH条件下展现优异HER性能进行机理探究。经X射线吸收光谱(XAS)研究发现在W1Mo1-NG中,异核W-Mo双金属原子锚定在N掺杂的石墨烯空位中,通过W-O和Mo-O键稳定并形成O配位W-Mo异核二聚体。W和Mo原子分别位于氧桥[WO4]四面体和畸变[MoO6]八面体单元中;W原子通过O原子与Mo原子桥接,构建双金属二聚体环境(图3A)。
Mo2-NG和W2-NG中Mo和W原子均处于八面体配位环境中,分别通过O和N原子锚定在NG上(图3B-C)。基于物理表征结果,建立了O-配位W-Mo异核双原子反应模型,为性能相关性的研究提供了结构基础。图3D所示W1Mo1-NG有六个非等价非金属活性位点,使其在全pH值下展现优异的H吸附性行为。
图3E-H所示电子在W1Mo1-NG体系中系统是离域的,这表明O配位的W-Mo异二聚体具有强的共价特性和弱的离子性质。相比于异核的W1Mo1-NG,同核Mo2-NG和W2-NG表现出强的离子性质(图3G,H),这使得金属中心的电子部分耗尽环绕配位的O位点,导致对H的过度结合。由于电子的离域,异核W1Mo1-NG的占据态从0.30 eV到EF(图6I中的阴影部分)的态密度(DOS)增大,导致良好的H吸附行为。
图6J所示在O配位的W1Mo1-NG和Mo2-NG系统中,O 2s和O 2pz价轨道与H 1s的杂化。总的来说,O-配位的W-Mo异二聚体比单元素体系具有较强的电子离域效应,使得W-Mo双原子体系对H具有更佳的吸附行为,电催化析氢反应动力学显著加快,最终提高了本征催化活性。独特的W-O-Mo-O-C结构与强共价相互作用,使得电子离域的W-Mo双原子催化剂在全pH范围具有良好的电催化析氢活性与稳定性,有望成为具有工业催化应用潜力的新型催化剂。该工作为从分子尺度认识催化反应的机理提供了理想的模型,为双金属原子催化的设计、机理研究提供了新思路。
近日,山西大学晶态材料研究所范修军教授和张献明教授在Science Advances上发表了题为“O-coordinated W-Mo dual-atom catalyst for pH-universal electrocatalytic hydrogen evolution”的文章(Sci. Adv., 2020, 6(23), eaba6586.)。
该研究工作对于大批量制备高活性的过渡金属双原子催化剂提供了一个借鉴,此外详细的表征与理论计算结合对于理解双原子催化剂的活性起源提供了更为深入的理解,为其他电催化催化剂的合成制备与机理探讨也有重要的指导意义。
山西大学晶态材料研究所博士研究生杨洋是本工作的第一作者,德州大学奥斯汀分校钱玉敏博士为本工作的共同第一作者。本工作得到国家自然科学基金、山西省高等学校中青年拔尖创新人才、山西省“1331工程”山西师范大学化学优势特色学科的资助,也得到了山西省“1331工程”创新团队和中科院山西煤化所煤转化国家重点实验室的支持。





